

【防控备用16】解读|如何避免出口防疫物资被召回、退货(防护服出口信息指南)
导 读
当前,我国已经成为全球抗疫医疗物资的主要生产供应国之一。医疗物资的质量安全监管问题,事关重大,高层始终高度重视。
以福州关区为例,开春以来,为缓解新冠防疫物资紧缺局面,部分服装家纺类企业在第一时间调整生产计划,着手新建防护服生产线支援抗疫一线,并很快开展对欧美等海外国家和地区的出口。
近期,随着防疫物资相关政策密集出台,为帮助企业稳定对外生产出口,迅速适应通关新形势、新要求,福州海关充分发挥技术性贸易措施工作职能,组织骨干力量奔赴生产一线,实地回应并帮助解决备案、送检、报关、查验等诸多问题。
如何才能让企业产品出口不再走弯路?为此,小编整理汇总了防护服出口的海关监管要点,供出口企业参考借鉴。
一、 国内对防护服产品如何分类


医用防护服是指医务人员(如医生、护士、公共卫生人员、清洁人员等)及进入特定区域的人群(如患者、医院探视人员、进入感染区域的人员等)所使用的防护性服装;大多采用PP(聚丙烯)、SMS无纺布、高密度聚乙烯材料,外覆专用透气膜的医疗防护服具有很好的防护性能,并且无毒无刺激性,对皮肤无害。需要满足防护性、舒适性、物理机械性能以及其它性能的要求。
国内医用防护服标准
1.GB/T 20097-2006 防护服一般要求
2.GB 19082-2009 医用一次性防护服技术要求
3.YY/T 1498-2016 医用防护服的选用评估指南
4.YY/T 1499-2016 医用防护服的液体阻隔性能和分级
5.YY/T 1632-2018 医用防护服材料的阻水性:冲击穿透测试方法
6.YY/T 0506-2016 病人、医护人员和器械用手术单、手术衣和洁净服
7.YY/T 0689-2008 血液和体液防护装备防护服材料抗血液传播病原体穿透性能测试 Phi-X174噬菌体试验方法
其中YY/T 1499-2016《医用防护服的液体阻隔性能和分级》中将医用防护服一共分为了4级,等级越高,防护性能越好。
其他应用领域防护服标准
1.GB 24539-2009《防护服装化学防护服通用技术要求》-工业防护服标准
2.GB 24540-2009《防护服装酸碱类化学品防护服》-工业防护服标准
3.GJB 2063-94《隔绝式防毒衣通用规范》-军用防护服标准
4.GJB 1971-94《FFY03型防毒衣规范》-军用防护服标准
5.GJB 1750-93《含碳透气防毒服通用规范》-军用防护服标准
注意:防护服/医用防护服是否合乎标准,需要经过检测机构检验。获认可的检测和校准实验室名录可登录https://las.cnas.org.cn/LAS_FQ/publish/externalQueryL1.jsp进行查询。
二、报关过程注意事项
1.医用防护服属于二类医疗器械,根据商务部/海关总署/国家药品监督管理局公2020年第5号(2020年3月31日),出口5类医疗物资(新型冠状病毒检测试剂、医用口罩、医用防护服、呼吸机、红外体温计)的企业向海关报关时,提交医疗器械产品注册证和企业承诺声明(新冠病毒检测试剂还须提供药监部门出具的出口销售证明)。
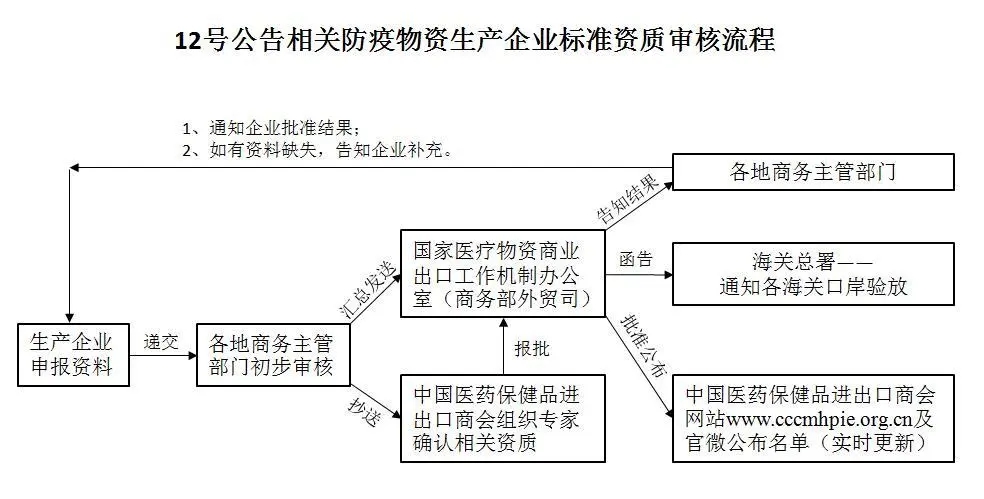
2.若产品(指上述5类医疗物资)取得国外标准认证或注册,根据商务部/海关总署/国家市场监督管理总局公告2020年第12号(2020年4月25日),出口企业在报关时须提交书面声明,承诺产品符合进口国(地区)质量标准和安全要求,海关凭商务部提供的取得国外标准认证或注册的生产企业清单(中国医药保健品进出口商会网站www.cccmhpie.org.cn动态更新)验放。


附:进入医保商会取得国外标准认证或注册的防疫物资生产企业清单流程(原则上每周报送一次,截止时间为每周三17:00。)
3.根据海关总署公告2020年第53号,自4月10日起,对“6210103010、3926209000”海关商品编号项下的2类医用防护服实施出口商品检验。
注意是医用防护服,重要的事情说三遍:“医用、医用、医用”。生产个人防护或者工业用非医疗器械管理的非医用防护服,未列入法检,也非5号、53号、12号公告所涉产品,有进出口权的企业,可自行直接出口,不要求提交注册证和质量安全承诺书;但企业要严格产品质量管控,坚持诚信经营、合规经营;在申报时,要如实填写医用/非医用、商品名称、规格型号等申报要素,否则一样被处罚。
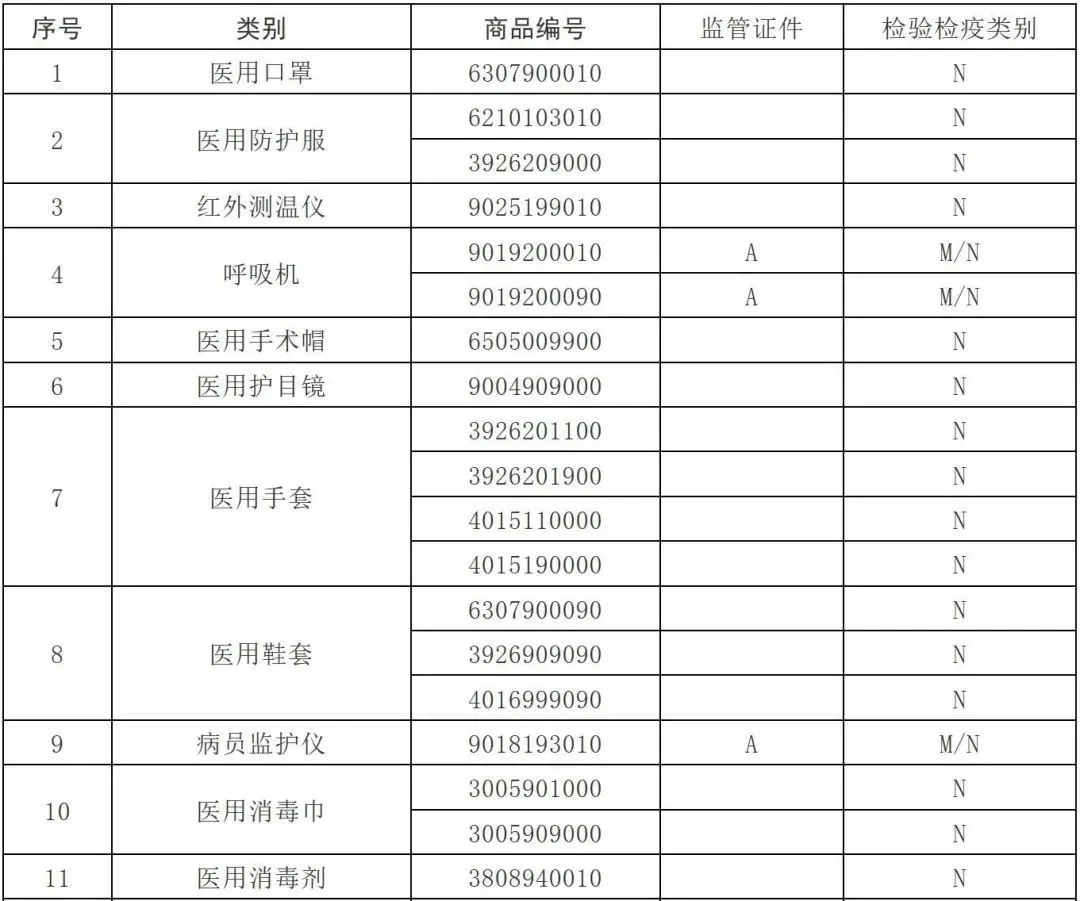
4.除特殊情况外,绝大部分医用防护服按照材质分类应归入下列税号:

5.防护服的出口退税率为13%。
6.中国已与25个国家和地区达成了17个自贸协定,自贸伙伴遍及欧洲、亚洲、大洋洲、南美洲和非洲;中国正与27个国家进行12个自贸协定谈判或者升级谈判,主要包括《区域全面经济伙伴关系协定》(RCEP)、中日韩、中国-挪威、中国-斯里兰卡、中国-以色列、中国-韩国自贸协定第二阶段、中国-巴基斯坦自贸协定第二阶段谈判,以及中国-新加坡、中国-新西兰自贸协定升级谈判等。中国原产的商品,包括疫情防控物资,出口到这些国家和地区凭优惠原产地证书可以获得关税减免优惠,以出口防护服到韩国为例(如下),建议出口前向海关申请原产地证书。
三、国外防护服准入条件
(一)美国
非医用防护服由美国国家职业安全卫生研究所(NIOSH)管理,企业直接在NIOSH官网进行注册申请;
医用防护服必须要取得美国食品和药物管理局FDA注册才可以在美国本土市场进行销售。
1.防护服分类
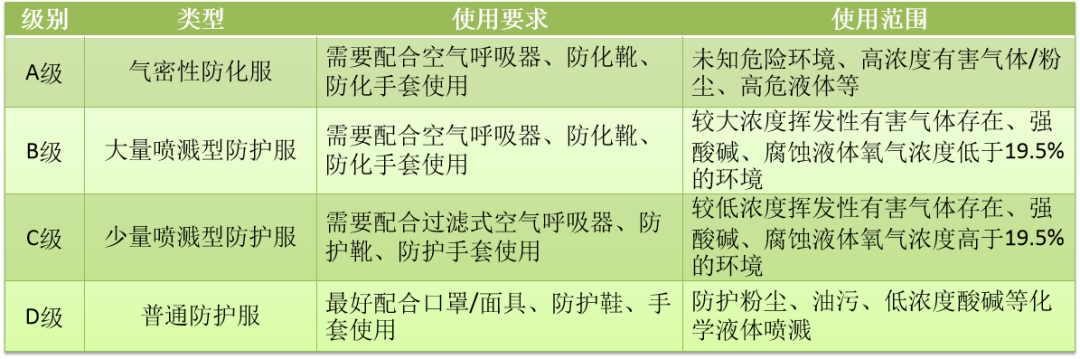
根据防护服不同应用,美国消防协会NFPA和美国材料实验协会ASTM提出并经美国职业安全和健康管理局OSHA认可,将防护服分为以下四个等级(NFPA1999:2018《紧急医疗行动的防护服和装备》):
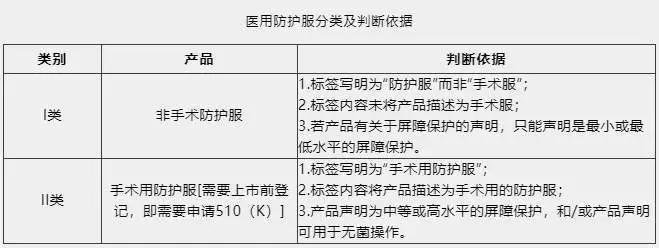
医用防护服可分为非手术防护服和手术用防护服两类。非手术防护服属于I类医疗器械,免于上市前登记,直接进行机构注册。而手术用防护服属于II类医疗器械,需要进行上市前登记,即需要申请FDA 510(K)。
2. 防护服标准
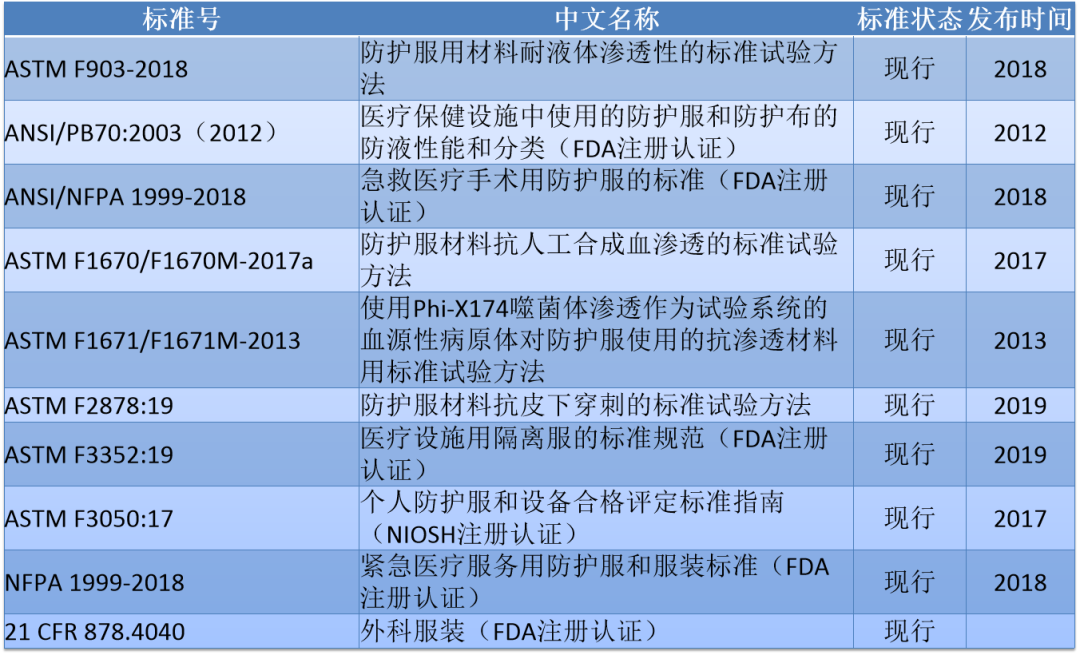
美国市面销售上的防护服也常用ANSI/AAMI PB70的4个级别对产品测试分类。各级别的阻隔性要求见下图:
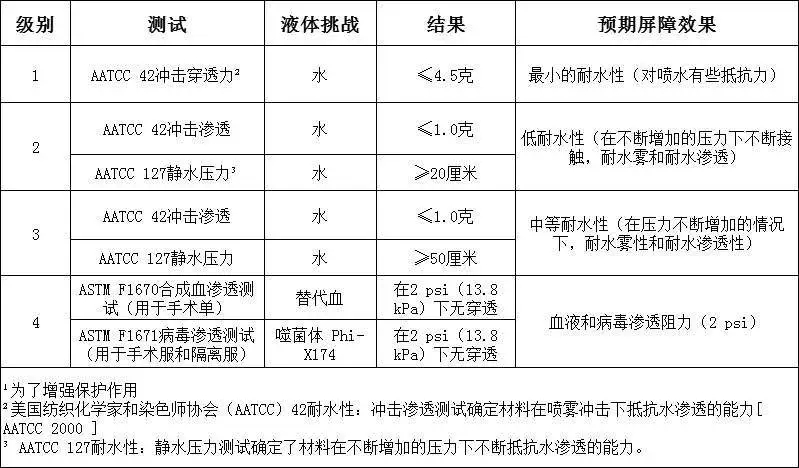
需要重点关注的是,级别1、2和3的要求具有与之相关的特定测试要求,仅对4级防护服进行了抗病毒渗透性测试,因此使用ASTM F1671仅将4级服装视为对病毒渗透不可渗透。符合较低级别(1、2和3级)的防护服不能被认为是不可渗透的,不能用在病原性防护中,只有美标4级才有能力用在此次新冠肺炎疫情的医用防护中。
3. FDA注册需要关注的问题点
问题一:FDA证书是哪个机构发放的?
答:FDA注册是没有证书的,产品通过在FDA进行注册,取得注册号码,FDA会给申请人一份回函(有FDA行政长官的签字),但不存在FDA证书一说。常所说的FDA认证就是指FDA注册号,而FDA也只认这个注册号。我们通常看到的FDA证书是中介代理机构(注册代理)签发给厂家,以证明其帮助该厂家完成了美国FDA要求的“生产设施注册和产品类型注册”(Establishment Registration and Device Listing),完成的标志是帮助厂家取得了FDA的注册登记号。

问题二:FDA需要指定的认证实验室检测吗?
答:FDA作为联邦执法机构,主要负责制定法规和市场监管等。FDA只会对服务性的检测实验室的GMP(英文Good Manufacturing Practice 的缩写,即“良好作业规范”)质量进行认可,合格的颁发合格证书,但不会向公众“指定”或推荐特定的一家或几家。

问题三:FDA注册是否一定需要一位美国代理人?
答:美国FDA规定,国外的医疗器械、食品、酒类、药品等工厂在进入美国之前必须进行注册,同时必须指定一位美国代理人,该美国代理人负责紧急情况和日常事务交流。美国代理人是指在美国或在美国有商业场所,国外工厂为了进行FDA注册而指定其为注册代理人。美国代理人不能只是邮箱、语音电话,或者作为国外工厂代理人的个人地址根本就不存在的场所。
问题四:FDA注册和FDA检测以及FDA认证三者到底有什么区别?
答:严格来讲没有FDA认证的说法。FDA批准一般针对药品比较多,就是允许这个药品上市了;FDA注册就是产品要先到FDA官网注册下,个别产品需要检测;FDA检测是针对与食品直接或间接接触的材料,由第三方根据FDA的公布的法规来做检测,看产品是否符合FDA法规要求。
问题五:FDA注册和CE认证有什么区别?
答:CE认证模式为:产品检测+报告证书,而FDA注册采用的是诚信宣告模式,即:你对自己的产品符合相关标准和安全要求负责,并在美国联邦网站注册,如果产品出事,那么就要承担相应的责任。因此FDA注册对于大部分产品,不存在寄样品检测和出证书的说法。
问题六:FDA注册有效期有多长?
答:FDA注册有效期为一年,如果超过一年,则需要重新提交注册,所涉及的年费也需要重新付。
问题七:如何查询产品已经获得FDA列名或510K注册?
答:唯一权威途径就是去FDA官网查询,FDA列名:https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfRL/rl.cfm

510K注册:
https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfPMN/pmn.cfm

(二)欧盟
非医用防护服须获得欧盟CE认证,并符合技术法规(EU)2016/425(PPE),该法规覆盖防护服、呼吸防护设备、手套等产品的安全监管。法规(EU)2016/425(PPE)原文网址如下:https://eur-lex.europa.eu/legal-content/EN/TXT/?qid=1584082251069&uri=CELEX:32016R0425
医用防护服按照医疗器械管理,其中无菌医用防护服需按照欧盟医疗器械指令93/42/EEC(简称MDD)或欧盟医疗器械条例EU2017/745(简称MDR)(注:MDR将于2020年5月26日期正式取代MDD获得CE认证),非无菌医用防护服只需进行CE自我声明。
1.欧盟医疗器械指令93/42/EEC(MDD)授权的公告机构查询地址:
https://ec.europa.eu/growth/tools-databases/nando/index.cfm?fuseaction="directive.notifiedbody&dir_id=13
2.欧盟医疗器械条例EU 2017/745(MDR)授权公告机构查询地址:
https://ec.europa.eu/growth/tools-databases/nando/index.cfm?fuseaction="directive.notifiedbody&dir_id=34"
3.欧盟个人防护装备条例EU2016/425(PPE)授权的公告机构查询地址:
https://ec.europa.eu/growth/tools-databases/nando/index.cfm?fuseaction="directive.notifiedbody&dir_id=155501
1.防护服分类
按照防护性能分为6类,从type1到type6,数字越小防护越高,type 4为医用推荐要求。
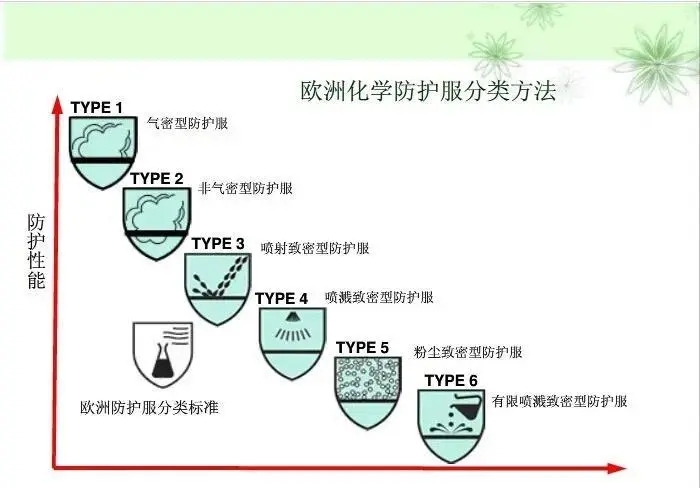
2.防护服标准及对应分类
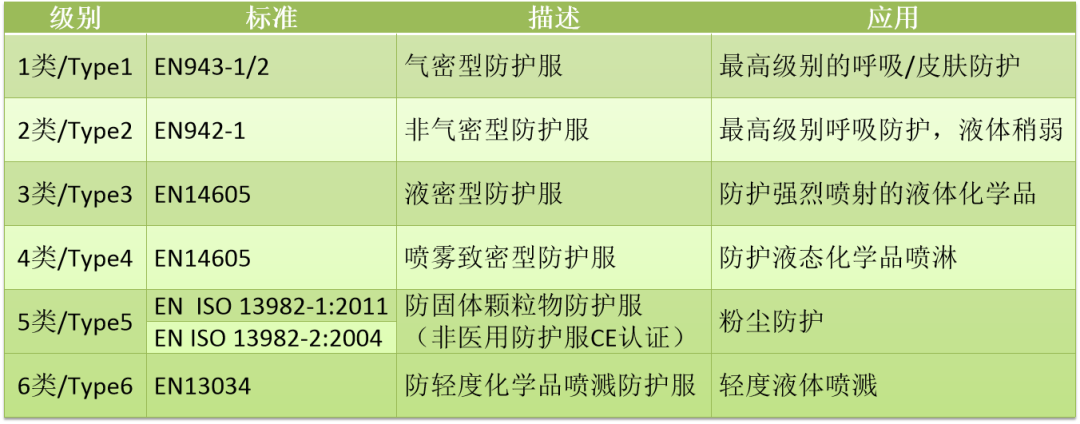
除上述标准之外,特别要说明的是EN 14126:2003+AC-2004 《防护服抗感染防护服的性能要求和试验方法中Type1至Type6的要求》,该标准适用于可重复的和有限使用的防护服;其中要求防护服的接缝处应符合EN 14325《化学药品防护服 化学防护服装材料、缝合线、联结和组合的试验方法和性能分类》中的强度要求。EN 13795-1:2019《外科服装和罩衫.要求和试验方法.第1部分.外科服装和罩衫》、EN 13795-2:2019《外科服装和窗帘.要求和试验方法.第2部分.洁净空气服》适合于外科医生及手术过程中为避免交叉感染的患者穿着用。
3.重点关注
根据抗击新冠疫情期间我国《国家卫生健康委办公厅关于加强疫情期间医用防护用品管理工作的通知》(国卫办医函〔2020〕98号),疫情防控期间,医用防护服不足时,医疗机构可使用紧急医用物资防护服。紧急医用物资防护服应当符合欧盟医用防护服EN14126标准(其中液体阻隔等级在2级以上)并取得欧盟CE认证,或液体致密型防护服(type3,符合EN14605标准)、喷雾致密型防护服(type4,符合EN14605标准)、防固态颗粒物防护服(type5,符合1SO13982-l&2标准)。Type 6 类防护服因为对接缝处没有技术要求,所以接缝和针孔处会造成污染液体的穿透,不适用于新型冠状病毒防护。紧急医用物资防护服仅用于隔离留观病区(房)、隔离病区(房),不能用于隔离重症监护病区(房)等有严格微生物指标控制的场所。

4.CE标志制度
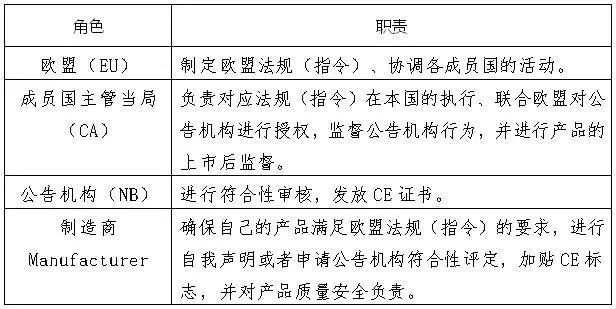
CE标志制度是欧盟对产品进入欧盟市场进行的监管方式。加贴CE标志的产品表明产品符合欧盟有关安全、健康、环保等法规要求,可以在欧盟27个成员国、欧洲贸易自由区的4个国家以及英国和土耳其合法上市销售。
按照欧盟规定,不同产品采用不同的评价方式加贴CE标志,主要有两种方式:绝大部分产品是制造商采取自我符合性声明方式,就可以加贴CE标志;部分风险相对更高的产品需要经过欧盟授权的第三方机构,即公告机构(Notified Body)进行符合性评定后,方可加贴CE标志。
下图列举了欧盟、成员国的主管当局、公告机构和制造商的职责。
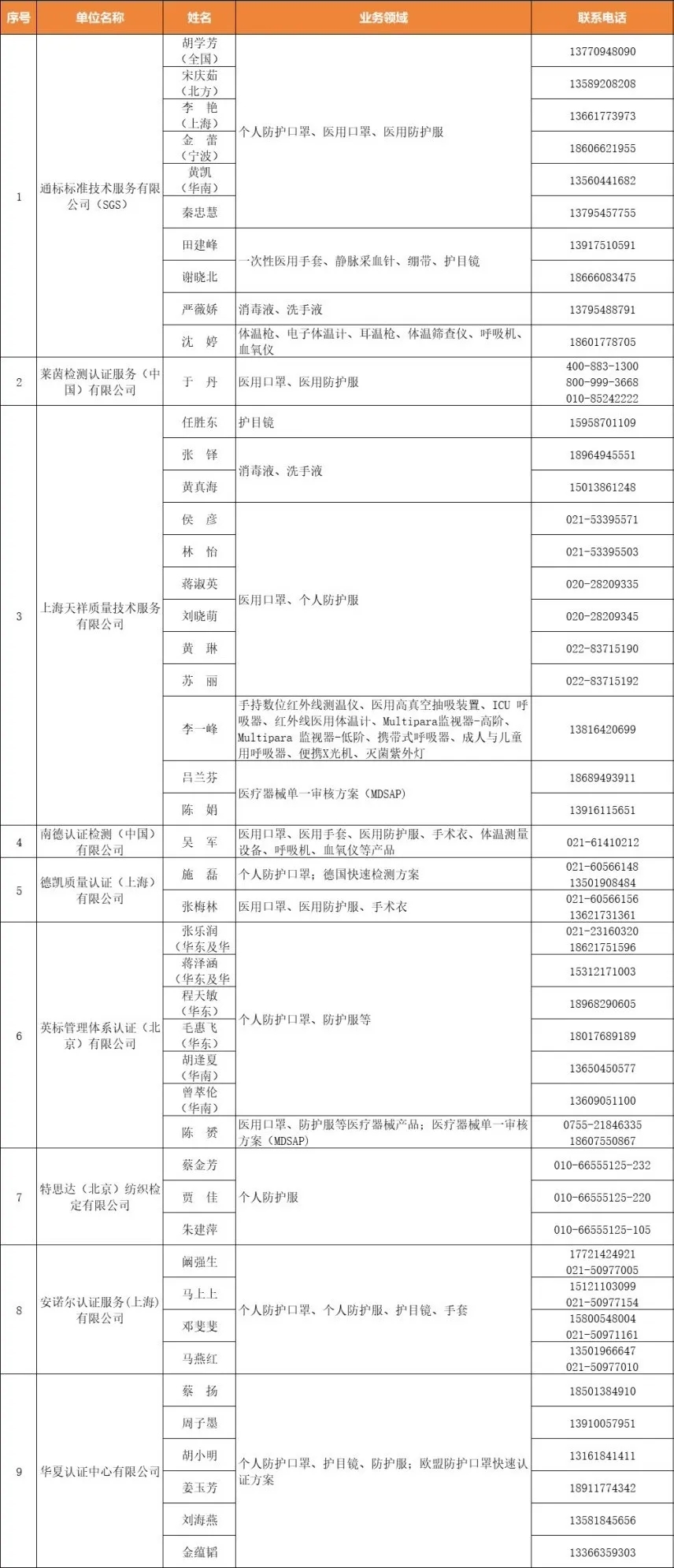
国内具备欧盟公告机构防护服等业务资质的认证机构名录(更新至2020年4月13日)
(三)日本
如果需要投放市场产品必须满足日本的Pharmaceutical and Medical Device Act(PMD Act《药品和医疗器械法》),在PMD Act的要求下,TOROKU注册系统要求国外的制造商必须向PMDA注册制造商信息。
日本的政府机构主要是制定法律法规和进行宏观管理,具体业务工作很多都交给各类行业协会来做。防护服涉及的主要行业协会为日本安全设备协会(JSAA)和日本防护服协议会(JPCA)。

● 日本安全设备协会(JSAA)网址:http://jsaa.or.jp/
● 日本医药品和医疗器械综合机构(PMDA)网址:www.pmda.go.jp
1.防护服技术标准
日本关于个体防护标准是归类于日本国家标准中的医疗安全用具T类标准的劳动安全范畴,其中JIS标准是日本国家级标准中最权威、最重要的标准,属于非强制性标准。但如被日本法律引用,JIS标准可成为强制性标准。

2.重点关注
JIS标准中涉及防护服的有45个标准,其中日本JIS T 8122:2015(预防危险生物制剂的防护服)将化学防护服标准与抗感染防护服标准相结合,其引用核心标准为JIS T 8115(化学防护服)、JIS T 8060(防止接触血液和体液的防护服-关于防护服材质对血液和体液的耐渗透性能的测定方法-使用人工血液的试验方法)、JIS T 8061(防止接触血液和体液的防护服-关于防护服材质对血液媒介性病原体的耐渗透性能的测定方法-使用Phi-X174噬菌体的试验方法)。故符合日本JIS T 8122:2015标准的防护服可适用于抗击新冠肺炎疫情的医疗和救治。
(四)韩国
韩国医疗器械准入的法规门槛,基本分类为I、II、III、IV类,持证为韩国公司(License holder),韩国收货人需要到韩国药监局Korea Pharmaceutical Traders Association(KPTA),提前备案进口资质,即由韩国本土企业向监管部门备案,网址:www.kpta.or.kr
1.防护服分类
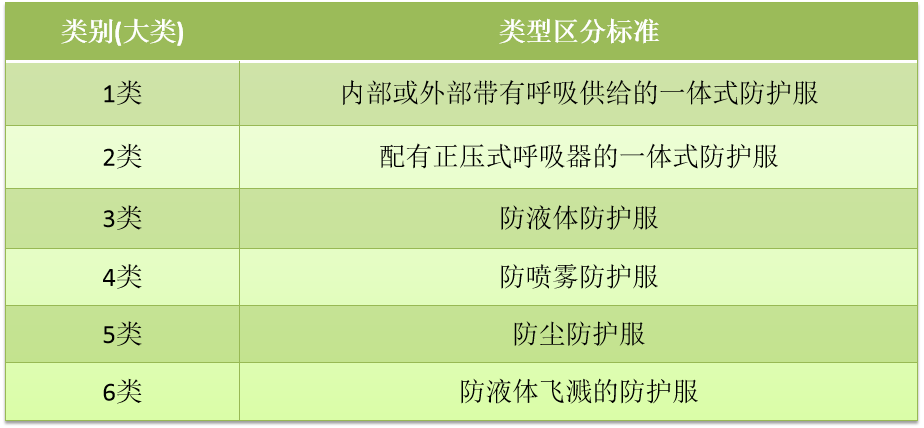
2020年1月15日,韩国劳动部2020-35号公告《防护设备安全认证通知》内给出了化学物质防护服、防尘口罩等性能标准和测试方法。根据《防护设备安全认证通知》内的防护服性能标准内容,防护服共分为六大类(其中第一类又分为5小类),类别见下图:
《防护设备安全认证通知》全文可在
http://www.law.go.kr/%ED%96%89%EC%A0%95%EA%B7%9C%EC%B9%99/%EB%B3%B4%ED%98%B8%EA%B5%AC%EC%95%88%EC%A0%84%EC%9D%B8%EC%A6%9D%EA%B3%A0%EC%8B%9C下载
2.防护服技术标准

(五)澳大利亚
须通过澳洲的TGA注册,TGA 是Therapeutic Goods Administration的简写,全称是治疗商品管理局。澳大利亚对医疗器械分为I类,Is and Im, IIa, IIb, III类,分别为豁免、备案和注册。无论哪类医疗器械,其上市销售前必须得到澳大利亚政府的准许,符合医疗器械的基本要求,按照符合性审查程序进行审查。
特别提醒:
澳大利亚已与欧盟达成互认协议。这意味着,合格评定证书由TGA颁发的也被欧盟认可,TGA也认可欧盟CE认证。已获CE认证的用户,可提交CE证书及相关资料,获得TGA证书。
如果产品已经注册或备案,制造商更换经销商对其没有影响。对国外产品进行注册审批后,每年还要常规注册一次,说明产品型号、性能及质量有无变化。TGA 全权负责对医疗器械的符合性评价,并收取一定费用,相关费用金额可参见 TGA的网站。
澳大利亚治疗商品管理局(TGA)官网网址:www.tga.gov.au
2002年澳大利亚颁布了《医疗器械法规2002>>,这是一部医疗器械管理方面的专门法规。TGA执行《治疗用品法案》、《医疗器械法规2002>>所赋予的产品市场准入和市场监管的职责,并保证在澳大利亚上市的医疗器械符合标准,并保证进一步地发展澳大利亚的治疗水平以及医疗器械工业。新的管理框架与 GHTF 的管理原则一致,于 2002 年 10月执行。澳大利亚 1998 年与欧盟的相互认可协定,使监管体系更加“协调” ,澳大利亚新的监管模式纳入欧盟的法规要求,然而不包括欧盟的第三方评价制度。虽然监管的主要内容被保留,但医疗器械分类和上市前评估要求有所改变,对上市后监督提出了具体的要求。
2002年《医疗器械法规》下载链接:https://www.legislation.gov.au/Series/F2002B00237
防护服技术标准

供稿单位:福州海关所属莆田海关
友情链接

地址:北京市朝阳区华严北里甲1号健翔山庄C11座
电子邮件:ccib@vip.163.com 联系电话: 010-82021949